The rate of evolution of the new coronavirus, and how the virus interacts with other microorganisms in the lungs of the infected, is still unclear.
Recently, the academic journal Clinical Infectious Diseases published an academic paper from the Chinese research team: "Genomic diversity of SARS-CoV-2 in Coronavirus Disease 2019 patients", focusing on the above issues.
The research team performed transcriptome sequencing of bronchoalveolar lavage fluid from 8 patients with new-type coronavirus pneumonia, 25 patients with community-acquired pneumonia (CAP), and parenchymal inflammation outside the hospital, and 20 healthy controls.
Studies have shown that elevated levels of virus diversity are found in some new coronavirus-infected individuals, suggesting that the virus is at risk for rapid evolution. Although no evidence of the spread of intra-host variants has been found, this risk should not be ignored.
The research team also said that the virus evolves in infected patients after infection, which may affect the virus's virulence, infectivity and spreadability.
Although it is unclear how intrahost mutations can spread among populations, it is necessary to strengthen monitoring of virus evolution and related clinical changes in populations.
It is worth noting that patients' new coronavirus genomes may be highly diverse, which is also observed in other viruses. High diversity may increase the adaptability of the virus population, making it difficult to eliminate.
Further research is needed to explore how this may affect the immune response to the virus and whether there are options for acting on different strains in the human body or during transmission.
The study concluded that, overall, the study revealed the evolution of a novel coronavirus in patients, a feature common to most RNA viruses.
How these mutations affect the fitness of the virus and the genetic diversity of the population requires further study.
Currently, public databases only share a limited number of sequences, so it is urgent to accumulate more sequences to track the evolution of the viral genome and link these changes to clinical symptoms and outcomes.
The thesis team members are from the following institutions: Beijing Institute of Genomics, Chinese Academy of Sciences; University of Chinese Academy of Sciences; Key Laboratory of National Health Commission, Institute of Pathogen Biology, Chinese Academy of Medical Sciences; Frontier Innovation Center for Biomedical Sciences, Peking University; Department of Critical Care Medicine; Guangdong Provincial Key Laboratory of Emerging Infectious Diseases; State Key Laboratory of Emerging Infectious Diseases; Fujian People's Hospital; First Affiliated Hospital of Xi'an Jiaotong University; Center for Crossover Excellence in Animal Evolution and Genetics at the Chinese Academy of Sciences.
The corresponding author of the thesis is Li Minghuan, a researcher and doctoral supervisor of the Beijing Institute of Genomics, Chinese Academy of Sciences.
High levels of intra-host variation in patients with new coronavirus infections
Based on current data, it is estimated that the basic number of early infections (R0) of new coronaviruses is between 2.2 and 3.5, posing a serious threat to public health.
Recent studies have identified bats as a possible source of the new coronavirus, which may use the same cell surface receptors as SARS-CoV, ACE2.
These studies have improved our understanding of new coronaviruses. However, our knowledge of new viruses is still limited.
In humans, new coronaviruses have to withstand strong immune pressure, which may cause the virus to accumulate mutations constantly, thus breaking the control of the immune system.
These mutations may cause changes in the virulence, infectivity and spread of the new coronavirus. Therefore, researchers believe that it is necessary to study the pattern and frequency of mutations.
The research team sequenced transcriptomes of bronchoalveolar lavage fluid (BALF) samples from eight patients with the new coronavirus infection. The 110 sequence studies collected between December 24, 2019 and February 9, 2020 found that the number of mutations within the host was between 0 and 51, with a median of 4, indicating that the virus has a high rate of evolution.
The research team identified a total of 84 in-host mutations with a minimum allele frequency (MAF, usually the frequency of an uncommon allele in a given population) greater than 5%, and a MAF of 25 mutations greater than 20%.
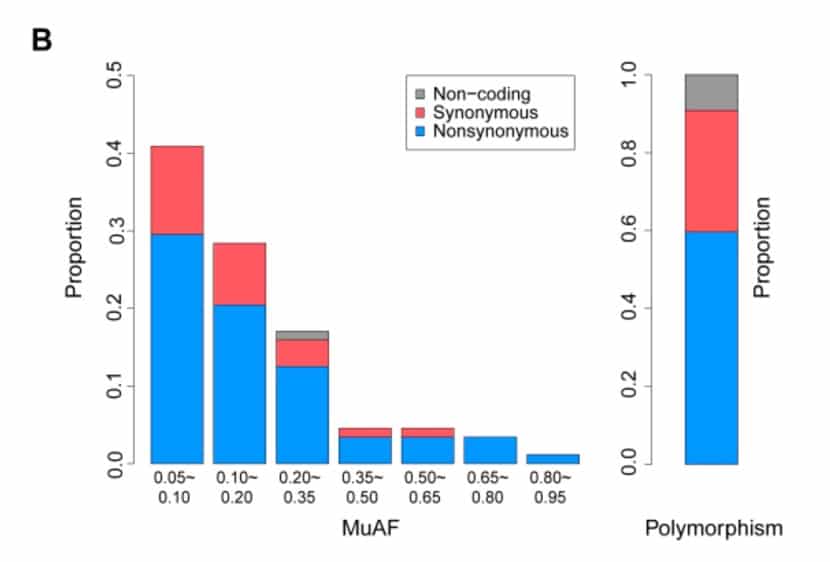
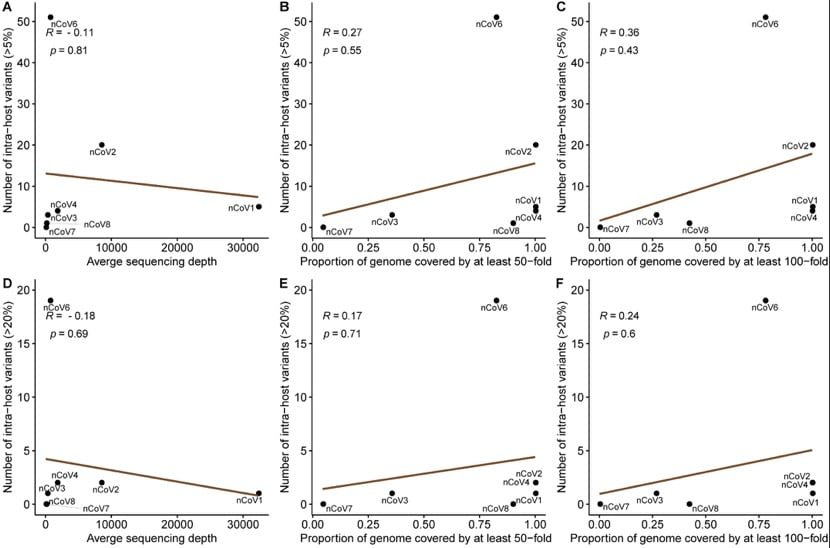
It is worth noting that the number of mutations is independent of the sequencing depth.
The overall Ka / Ks (the ratio between the synonymous substitution (Ka) and the synonymous substitution (Ks) can determine whether there is selection pressure acting on this protein-coding gene) is significantly less than 1, which is observed in the host variation and population data The resulting polymorphisms are similar, suggesting that purification selection affects both types of mutations.
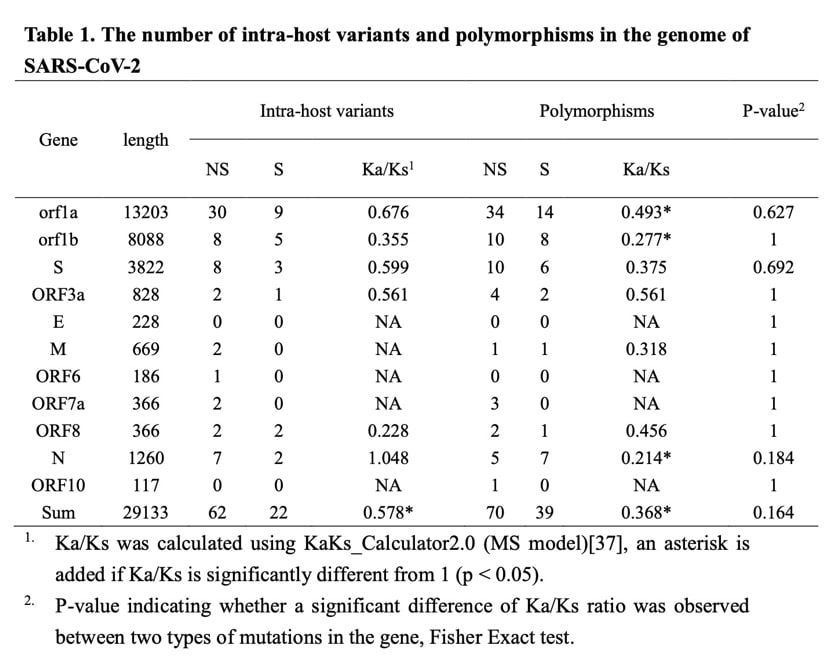
The number of variants observed in the gene is proportional to the length of the gene (for in-host variation, cor = 0.950, p = 8E-06; for polymorphisms, cor = 0.957, p = 4E-06). Although only a small number of mutations were observed in multiple patients (2 of 84 patients).
However, mutations are more likely to occur at certain locations, such as at position 10779. Mutant allele A was observed in all 7 patients, with frequencies ranging from 15% to 100%.
The number of variations within the host of each individual showed a large difference (MAF ≥5% variation was 0 to 51, with a median of 4; MAF ≥20% variation was 0 to 19, with a median of 1), This cannot be done with batch effects, coverage differences, or contamination (one to four of new coronavirus infections is nCoV1-4 in one batch, and five to eight of new coronavirus infections is nCoV5-8 in another batch; most mutations (Not observed in population data).
The research team also noticed that the amount of variation was not related to the number of days after the onset of symptoms or the age of the patient.
Overall, no cause was found to cause very high levels of variants in six of the new coronavirus infections (51 variants).
The research team says that a larger population is needed to study the frequency of such outliers and whether they are related to the level of host immune response or virus replication.
Researchers also noticed outliers from other viruses. It is worth noting that the source of the mutation may be a mutation that occurs in the body after infection, or it may be a new multi-spread coronavirus strain.
The research team mentioned during the discussion that RNA viruses have a high mutation rate. As a result, RNA viruses are prone to develop resistance to drugs and evade immune surveillance.
The mutation rate of the new coronavirus remains unclear. However, this study concluded that the mutation rate of the new coronavirus should be on the same order of magnitude as SARS-CoV. High mutation rates also lead to high levels of intra-host mutations in RNA viruses.
For variants with a frequency of 5% or more, the median variant in the host of patients with new-type coronavirus pneumonia was 4, which was not significantly different from the incidence reported in the Ebola virus disease study (frequency in 134 samples ≥ 5% of 655 variants, p> 0.05), which indicates that the mutation rate of the new coronavirus is also comparable to that of Ebola virus.
In addition, previous research has proposed that an exonuclease (ExoN) can provide corrective activity in SARS-CoV, and noted that all three key motifs in this gene are between SARS-CoV and the novel coronavirus. identical.
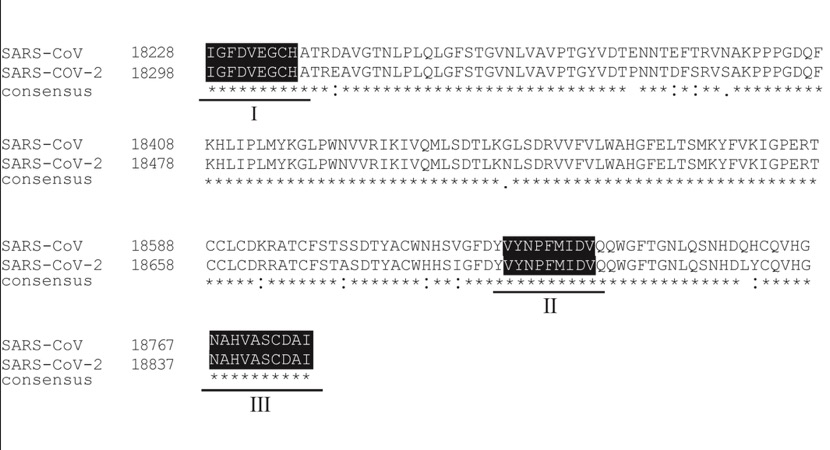
In addition, no polymorphism or intra-host variation was detected in any of these motifs, suggesting that the gene is highly conserved and therefore it may be a potential target for antiviral therapy.
The research team pointed out that although no mutation hotspot genes (base sequences with a higher probability of mutation) were found in the polymorphisms or in-host mutations, a common in-host mutation was observed among different individuals.
This implies the possibility of adaptive evolution of the virus in patients, which may affect its antigenicity, virulence and virus infectivity.
It is worth noting that patients' new coronavirus genomes may be highly diverse, which is also observed in other viruses. High diversity may increase the adaptability of the virus population, making it difficult to eliminate.
Further research is needed to explore how this may affect the immune response to the virus and whether there are options for acting on different strains in the human body or during transmission.
No evidence of the spread of internal host variation between samples
Of the eight new coronavirus-infected individuals, four (nCoV4) and seven (nCoV7) were from the same family, and the dates of onset of symptoms differed by five days.
It is highly suspected that the virus was transmitted from the infected four to the infected seven, especially considering that only the infected four had entered the Wuhan South China Sea product market, which was the starting point of the outbreak and was suspected to be the origin of the new coronavirus.
First, for two samples, the consensus sequence of the virus was the same, and all four in-host variants that passed the selection criteria for infected four were not detected in infected seven.
The researchers further expanded the scope of the study to all mutations with MAF ≥ 2% and were supported by at least 3 reads. By doing so, the researchers detected a total of 7 mutations (25 in total) between the two samples.
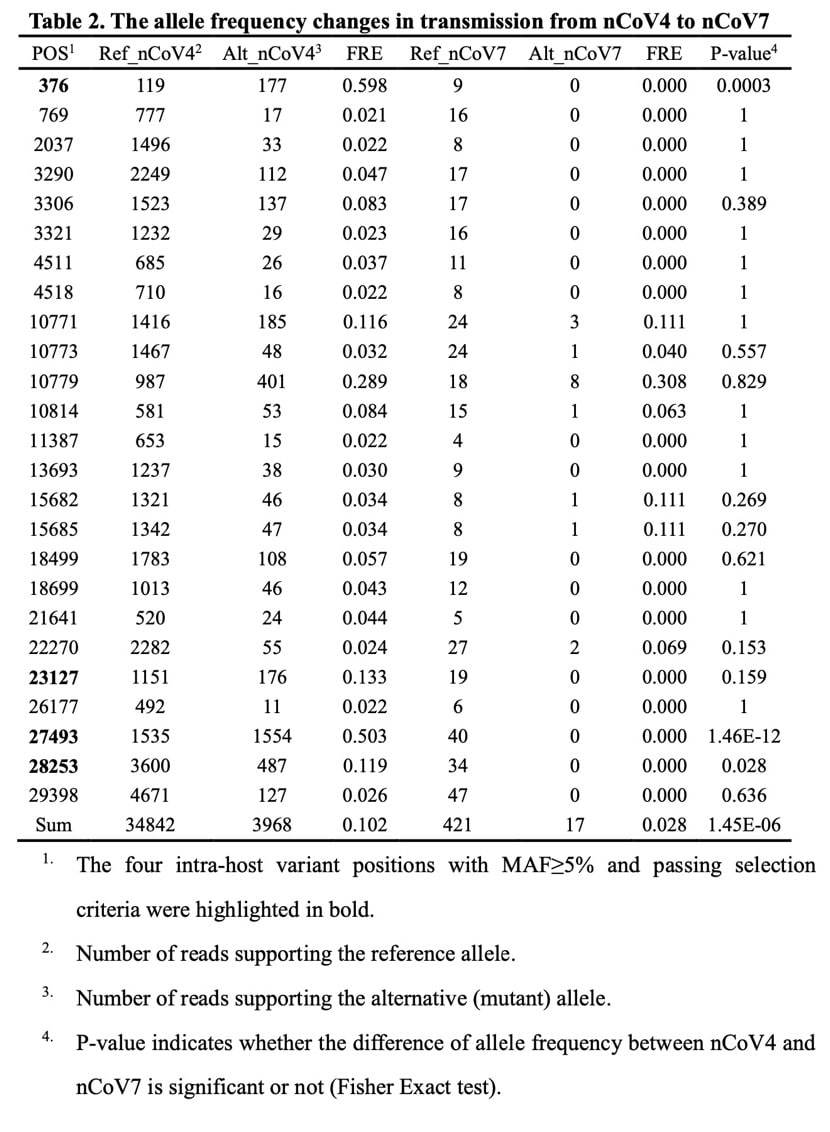
However, MAF in infected four and infected seven was similar to MAF in other samples, suggesting that these positions are either error-prone or mutated. Therefore, they cannot support the spread of these mutations.
At the same time, out of all 84 host internal mutations, only 3 were polymorphic in the population data (positions 7866 G / T; 27493 C / T; 28253 C / T).
This small overlap also indicates that intra-host variation is rarely transmitted to other samples. However, the possibility that the current database underestimates sequence diversity in the population cannot be ruled out.
The research team also mentioned in the discussion that during the single transmission event of the study, the research team did not find evidence of transmission of multiple strains.
However, it is unclear whether these intra-host mutations occur before or after transmission, and this will lead to different conclusions.
In addition, transmission may encounter bottlenecks, which may also lead to a loss of diversity.
No specific microbiome pattern found in people with new coronavirus infection
In addition to pathogens, other microbiota in the lungs are also related to the susceptibility and severity of the disease. Changes in the lung microbiota may alter immune responses to viral and secondary bacterial infections.
Therefore, understanding the microbiome, including bacteria that may cause secondary infections or affect the mucosal immune system, may help predict and reduce complications.
The mucosal immune system refers to the lymphatic tissue widely distributed in the respiratory tract, gastrointestinal tract, urogenital tract, and some exocrine glands. It is the main site for performing local specific immune functions.
Overall, there were significant differences in the composition of the microbiota between patients with new coronavirus infection, patients with community-acquired pneumonia, and the healthy group (R2 = 0.07, p = 0.001).
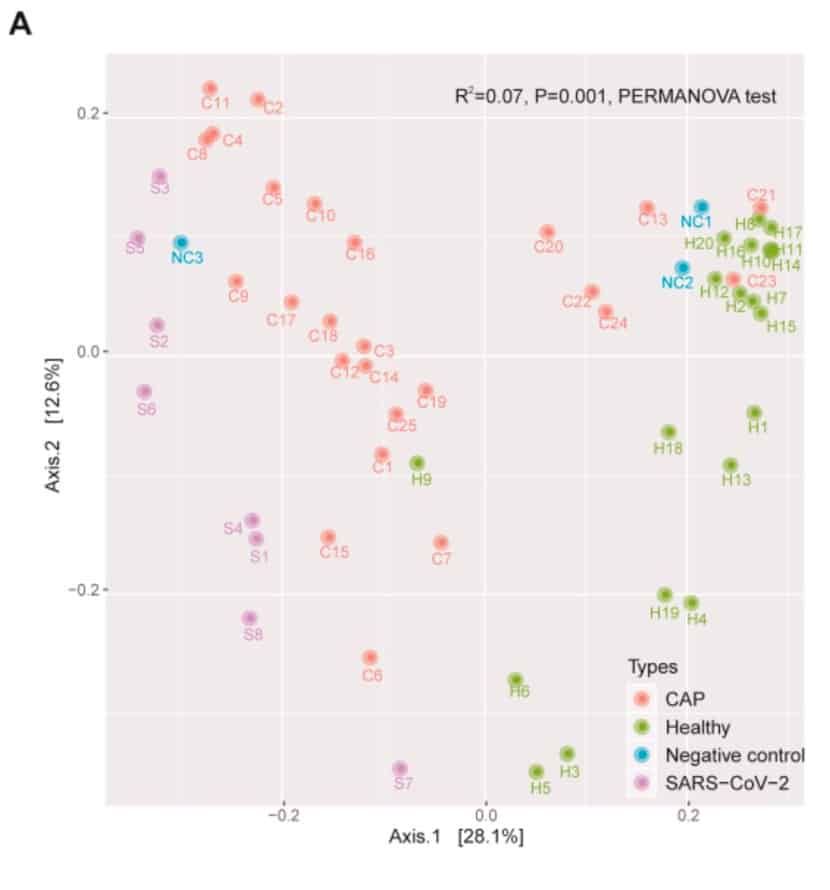
However, cluster analysis of samples of some new coronavirus-infected individuals shows that the microbiome of some samples is relatively poor.
After removing problematic samples, the study still found that patients with new coronavirus infection and patients with community-acquired pneumonia were different from healthy controls (new-corovirus infection and health group: R2 = 0.45, p = 0.001; patients with community-acquired pneumonia And healthy group: R2 = 0.10, p = 0.002), which indicates that their lung flora has a microecological disorder.
The microbiome can be divided into three different types. In particular, the microbiota in cluster I is mainly caused by possible pathogens, while the microbes in other clusters are more diverse.
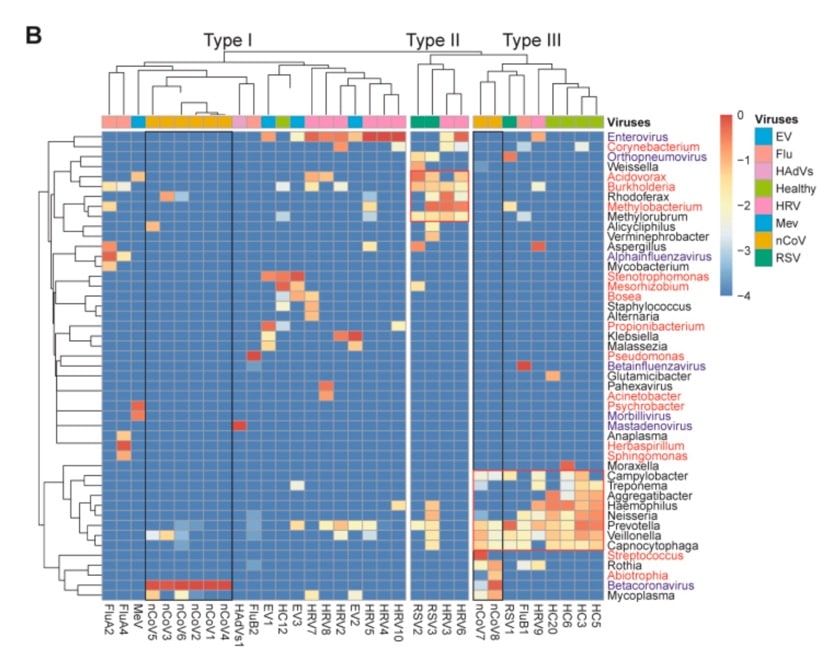
By further investigating the type of each cluster, the research team found that type III bacteria are mainly symbiotic bacteria often observed in the oral cavity and respiratory tract, while type II bacteria are mainly environmental microorganisms, so it is highly suspected that it is actually contaminated.
Therefore, the microbiota is either rich in pathogens (type I) or commensal bacteria (type III), or cannot be determined due to low microbial load (type II).
Of the eight infected individuals tested, the microbiota in samples of six new coronavirus infected individuals was pathogen-rich, while the other two samples were rich in symbiotic bacteria.
In addition, samples of new coronavirus-infected patients (infected two and infected six) with too many variants of new coronaviruses in both hosts were pathogen-enriched.
The paper mentions that the virus's overwhelming advantage may be associated with higher replication rates and may also potentially stimulate a strong immune response against the virus, in which case excessive intra-host mutations are expected.
However, as the analysis included only eight individuals infected with the new coronavirus, and the absolute microbial load was unknown, further data are needed for further research.
The researchers discussed that, except that the pneumonia group had significantly lower microbial diversity than the healthy control group, they did not find any specific microbiome patterns in patients with the new coronavirus pneumonia, nor in patients with CAP.
One possible cause may be the use of antibiotics in patients with pneumonia. However, this does not apply to all pneumonia samples, as a significant proportion of bacteria were found in some samples, including two patients with the new coronavirus pneumonia.
It is currently known that secondary infections of viral infections, especially respiratory viruses, often lead to a significant increase in morbidity.
Therefore, elevated levels of bacteria in BALF in some patients with new-type coronavirus pneumonia may increase the risk of secondary infection. In clinical data, the secondary infection rate of patients with new coronavirus pneumonia is between 1% and 10%.
However, the quantitative relationship between bacterial relative abundance / titer and infection is unclear.
Original article link:
https://academic.oup.com/cid/advance-article/doi/10.1093/cid/ciaa203/5780800?searchresult=1#
Special Report: Fighting The New Coronavirus